quicksand-build
Description
To run quicksand, an underlying datastructure with databases and reference genomes is required. This datastructure consists of:
A preindexed Kraken-database
A directory containing the indexed
fastafiles of the mt-genomes from RefSeq that were used to create the kraken-databaseIn the same directory, a file
taxid_map.tsv, linking the files in the folder to NCBI Taxonomy IdsA directory containing a
bed-file for each genome, indicating non-informative and low-complexity regions within the genome
Instead of creating this files manually, the supplementary pipeline quicksand-build is used for that. Note that quicksand-build is a separate pipeline hosted here.
Workflow
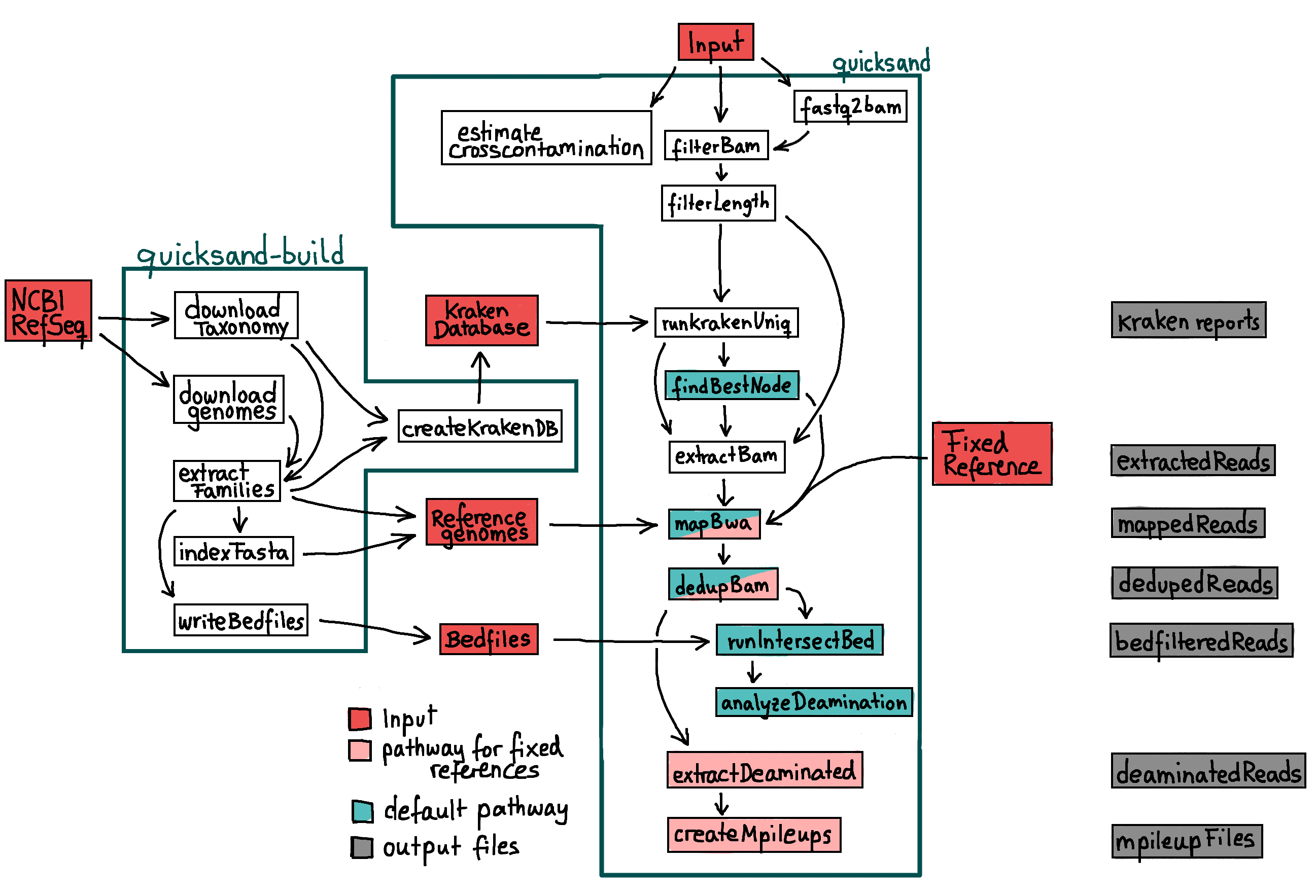
downloadTaxonomy / downloadGenomes
The first step of the pipeline is downloading of the genbank-files from the `NCBI RefSeq mitochondrion FTP-Server<https://ftp.ncbi.nlm.nih.gov/refseq/release/mitochondrion/>`_ and
the NCBI taxonomy using the kraken-build --download-taxonomy command.
extractFamilies
A custim python script is used to extract the fasta-files from the NCBI genbank files. Use the --include flag to specify taxa that
should be extracted. (default: all). All included taxa are used to build the kraken-database and can thus be identified by the quicksand pipeline.
indexFasta
The extracted fasta files are indexed using the bwa index command.
writeBedfiles
- For each
fastafile,dustmaskeris used to specify non-informative and low-complexity regions and write them as coordinates into a separate
bedfile.
createKrakenDB
Use kraken-build to add the extracted fasta files to the database. The database is then built using the with the flag --kmers specified kmer sizes
(default: 22)
Usage
Note
-profile docker flag to use docker instead.To run the pipeline with default parameters open the terminal and type:
nextflow run mpieva/quicksand-build --outdir <PATH> [ --kmers KMERS --include TAXA --exclude PATH ]
Flags
Flag |
Input type |
Description |
|---|---|---|
--outdir |
STRING |
Specify the directory
used to save the datastructure in
Default: 'out'
|
--include |
STRING |
A string of comma-separated taxa (exact match to the ncbi taxonomy names)
that should be included in the kraken database
Default: 'root' (all)
Example:
--include Mammalia,Aves
|
--kmers |
STRING |
A string of comma-separated numbers.
For each number a kraken database is built with the respective kmer-size
Default: '22'
outdir
├── kraken
│ ├── Mito_db_kmer22
│ ├── Mito_db_kmer23
│ └── Mito_db_kmer24
Example:
--kmers 22,23,24
|
--exclude |
PATH |
A TSV file containing family and comma-separated species names.
| The listed species are excluded from the kraken database
input (exclude.tsv):
Hominidae Homo_sapiens,Homo_neandertalensis
Bovidae Capra_aegagrus
Example:
--exclude exclude.tsv
|
Output
The output of quicksand-build is structured as follows:
outdir
├── kraken
│ ├── Mito_db_kmer22
│ │ ├── taxonomy
│ │ ├── ...
│ │ └── database.kdb
│ └── Mito_db_kmer24
│ ├── taxonomy
│ ├── ...
│ └── database.kdb
├── genomes
│ ├── ${Family}
│ │ ├── ${Species}.fasta
│ │ ├── ${Species}.fasta.fai
│ │ └── ...
│ └── taxid_map.tsv
├── masked
│ └── ${Species}.masked.bed
├── ncbi
│ └── raw gbff.gz files
└── work
└── intermediate nextflow files
The taxid_map.tsv file contains the following information:
1425170 Hominidae Homo_heidelbergensis Primates
9605 Hominidae Homo_heidelbergensis Primates
9604 Hominidae Homo_heidelbergensis Primates
9443 Hominidae Homo_heidelbergensis Primates
63221 Hominidae Homo_sapiens_neanderthalensis Primates
9605 Hominidae Homo_sapiens_neanderthalensis Primates
9604 Hominidae Homo_sapiens_neanderthalensis Primates
9443 Hominidae Homo_sapiens_neanderthalensis Primates
9606 Hominidae Homo_sapiens Primates
9605 Hominidae Homo_sapiens Primates
9604 Hominidae Homo_sapiens Primates
9443 Hominidae Homo_sapiens Primates
741158 Hominidae Homo_sapiens_subsp._'Denisova' Primates
9605 Hominidae Homo_sapiens_subsp._'Denisova' Primates
9604 Hominidae Homo_sapiens_subsp._'Denisova' Primates
9443 Hominidae Homo_sapiens_subsp._'Denisova' Primates
9605 Hominidae Homo_heidelbergensis Primates
9605 Hominidae Homo_sapiens_neanderthalensis Primates
9605 Hominidae Homo_sapiens Primates
9605 Hominidae Homo_sapiens_subsp._'Denisova' Primates
taxid_map.tsv file is used to provide the 'mapBwa' processNote
In case of editing or manually building the datastructure, be aware that the $\{Species\} in the taxid_map.tsv file must correspond to
the $\{Species\} filenames in the genomes and masked directories. Otherwise quicksand won't find the appropriate files